Metabolic network modeling with COBRA#
Databases of models#
Documentation of cobrapy and cobra toolbox#
Solvers#
To install cobrapy, run the following code below (requires pip):
# install cobrapy
# !pip install cobra
# !pip install pytest
# load modules
import cobra
from cobra.io import load_model
# check cobra version
cobra.__version__
'0.26.3'
model = load_model("textbook")
model
| Name | e_coli_core |
| Memory address | 7fcd71b2b610 |
| Number of metabolites | 72 |
| Number of reactions | 95 |
| Number of genes | 137 |
| Number of groups | 0 |
| Objective expression | 1.0*Biomass_Ecoli_core - 1.0*Biomass_Ecoli_core_reverse_2cdba |
| Compartments | cytosol, extracellular |
You can also load a exist model by using io functions like load_matlab_model, read_sbml_model …#
## io functions
# cobra.io.read_sbml_model
# cobra.io.load_json_model
# cobra.io.load_matlab_model
# cobra.io.load_yaml_model
Components of model#
Model (cobra.core.model.Model)#
attribute:#
| Attribute | Type | Description |
|---|---|---|
| reactions | DictList | A DictList where the key is the reaction identifier and the value a Reaction |
| metabolites | DictList | A DictList where the key is the metabolite identifier and the value a Metabolite |
| genes | DictList | A DictList where the key is the gene identifier and the value a Gene |
| solution | cobra.Solution | The last obtained solution from optimizing the model. |
| boundary | DictList | Boundary reactions in the model. Reactions that either have no substrate or product. |
| exchanges | DictList | Exchange reactions in model. Reactions that exchange mass with the exterior. Uses annotations and heuristics to exclude non-exchanges such as sink reactions. |
| demands | DictList | Demand reactions in model. Irreversible reactions that accumulate or consume a metabolite in the inside of the model. |
| sinks | DictList | Sink reactions in model. Reversible reactions that accumulate or consume a metabolite in the inside of the model. |
| objective | optlang.interface.Objective | Before introduction of the optlang based problems, this function returned the objective reactions as a list. With optlang, the objective is not limited a simple linear summation of individual reaction fluxes, making that return value ambiguous. Henceforth, use cobra.util.solver.linear_reaction_coefficients to get a dictionary of reactions with their linear coefficients (empty if there are none) The set value can be dictionary (reactions as keys, linear coefficients as values), string (reaction identifier), int (reaction index), Reaction or problem.Objective or sympy expression directly interpreted as objectives. |
methods:#
| Method | Parameters | Return type | Description |
|---|---|---|---|
| copy | None | cobra.Model | Provides a partial ‘deepcopy’ of the Model. All of the Metabolite, Gene, and Reaction objects are created anew but in a faster fashion than deepcopy |
| add_metabolites | metabolite_list (list) | None | Will add a list of metabolites to the model object and add new constraints accordingly. |
| remove_metabolites | metabolite_list (list) destructive (bool) |
None | Remove a list of metabolites from the the object. |
| add_boundary | metabolite (cobra.Metabolite) type (str, {"exchange", "demand", "sink"}) reaction_id (str, optional) lb (float, optional) ub (float, optional) sbo_term (str, optional) |
cobra.Reaction | Add a boundary reaction for a given metabolite. There are three different types of pre-defined boundary reactions: exchange, demand, and sink reactions. An exchange reaction is a reversible, unbalanced reaction that adds to or removes an extracellular metabolite from the extracellular compartment. A demand reaction is an irreversible reaction that consumes an intracellular metabolite. A sink is similar to an exchange but specifically for intracellular metabolites. If you set the reaction type to something else, you must specify the desired identifier of the created reaction along with its upper and lower bound. The name will be given by the metabolite name and the given type. |
| add_reactions | reaction_list (list) | None | Add reactions to the model. Reactions with identifiers identical to a reaction already in the model are ignored. |
| remove_reactions | reaction_list (list) remove_orphans (bool) |
None | Remove reactions from the model. |
| slim_optimize | error_value (float, None) message (string) |
float | Optimize model without creating a solution object. |
| optimize | objective_sense ({None, 'maximize' 'minimize'}, optional) raise_error (bool) |
cobra.Solution | Optimize the model using flux balance analysis. |
| summary | solution (cobra.Solution, optional) threshold (float, optional) fva (pandas.DataFrame or float, optional) names (bool, optional) float_format (callable, optional) |
cobra.ModelSummary | Create a summary of the exchange fluxes of the model. |
# components of a cobra Model
print(f"{len(model.reactions)} Reactions in the model\n------------------")
for r in model.reactions[:10]:
print(f"{r.id}: {r.reaction}, associated with {r.gene_name_reaction_rule}")
print()
print(f"{len(model.metabolites)} Metabolites in the model\n------------------")
for m in model.metabolites[:10]:
print(f"{m.id}: {m.name}")
print()
print(f"{len(model.genes)} Genes in the model\n------------------")
for g in model.genes[:10]:
print(f"{g.id}: {g.name}")
95 Reactions in the model
------------------
ACALD: acald_c + coa_c + nad_c <=> accoa_c + h_c + nadh_c, associated with mhpF or adhE
ACALDt: acald_e <=> acald_c, associated with G_s0001
ACKr: ac_c + atp_c <=> actp_c + adp_c, associated with ackA or tdcD or purT
ACONTa: cit_c <=> acon_C_c + h2o_c, associated with acnB or acnA
ACONTb: acon_C_c + h2o_c <=> icit_c, associated with acnB or acnA
ACt2r: ac_e + h_e <=> ac_c + h_c, associated with
ADK1: amp_c + atp_c <=> 2.0 adp_c, associated with adk
AKGDH: akg_c + coa_c + nad_c --> co2_c + nadh_c + succoa_c, associated with sucA and lpd and sucB
AKGt2r: akg_e + h_e <=> akg_c + h_c, associated with kgtP
ALCD2x: etoh_c + nad_c <=> acald_c + h_c + nadh_c, associated with adhP or frmA or adhE
72 Metabolites in the model
------------------
13dpg_c: 3-Phospho-D-glyceroyl phosphate
2pg_c: D-Glycerate 2-phosphate
3pg_c: 3-Phospho-D-glycerate
6pgc_c: 6-Phospho-D-gluconate
6pgl_c: 6-phospho-D-glucono-1,5-lactone
ac_c: Acetate
ac_e: Acetate
acald_c: Acetaldehyde
acald_e: Acetaldehyde
accoa_c: Acetyl-CoA
137 Genes in the model
------------------
b1241: adhE
b0351: mhpF
s0001: G_s0001
b3115: tdcD
b1849: purT
b2296: ackA
b1276: acnA
b0118: acnB
b0474: adk
b0116: lpd
# deepcopy a cobra model
model_copy = model.copy()
new_reaction = cobra.Reaction("new_rxn_1")
model_copy.add_reactions([new_reaction])
len(model_copy.reactions), len(model.reactions)
(96, 95)
# model objective
print(model.objective)
Maximize
1.0*Biomass_Ecoli_core - 1.0*Biomass_Ecoli_core_reverse_2cdba
# check the reaction in the objective function
model.reactions.get_by_id("Biomass_Ecoli_core")
| Reaction identifier | Biomass_Ecoli_core |
| Name | Biomass Objective Function with GAM |
| Memory address | 0x7fcd71766850 |
| Stoichiometry |
1.496 3pg_c + 3.7478 accoa_c + 59.81 atp_c + 0.361 e4p_c + 0.0709 f6p_c + 0.129 g3p_c + 0.205 g6p_c + 0.2557 gln__L_c + 4.9414 glu__L_c + 59.81 h2o_c + 3.547 nad_c + 13.0279 nadph_c + 1.7867 oaa_c... 1.496 3-Phospho-D-glycerate + 3.7478 Acetyl-CoA + 59.81 ATP + 0.361 D-Erythrose 4-phosphate + 0.0709 D-Fructose 6-phosphate + 0.129 Glyceraldehyde 3-phosphate + 0.205 D-Glucose 6-phosphate + 0.2557... |
| GPR | |
| Lower bound | 0.0 |
| Upper bound | 1000.0 |
Reactions (cobra.core.reaction.Reaction)#
attribute:#
| Attribute | Type | Description |
|---|---|---|
| id | string | The identifier to associate with this reaction |
| name | string | A human readable name for the reaction |
| reaction | string | Human readable reaction string |
| lower_bound | float | The lower flux bound |
| upper_bound | float | The upper flux bound |
| bounds | tupple | The upper and the lower flux bounds |
| subsystem | string | Subsystem where the reaction is meant to occur |
| objective_coefficient | float | The coefficient for this reaction in a linear objective |
| flux_expression | sympy expression | Forward flux expression |
| flux | float | The flux value in the most recent solution. |
| reduced_cost | float | The reduced cost in the most recent solution. |
| metabolites | dict (cobra.metabolite: float) | The dict of metabolites involved in the reaction. |
| genes | frozenset (cobra.gene) | The frozenset of genes involved in the reaction. |
| gene_reaction_rule | string | The boolean representation of the gene requirements for the reaction to be active. |
methods:#
| Method | Parameters | Return type | Description |
|---|---|---|---|
| remove_from_model | remove_orphans=False (bool) | None | Removes the reaction from a model. |
| get_coefficient | metabolite_id (str or cobra.Metabolite) | float | Return the stoichiometric coefficient of a metabolite. |
| get_coefficients | metabolite_ids (iterable) | map | Return the stoichiometric coefficients for a list of metabolites. |
| add_metabolites | metabolites_to_add (dict)
combine (bool) reversibly (bool) |
None | Add metabolites and stoichiometric coefficients to the reaction. If the final coefficient for a metabolite is 0 then it is removed from the reaction. |
| subtract_metabolites | metabolites (dict) combine (bool) reversibly (bool) |
None | Subtract metabolites from a reaction. That means add the metabolites with -1*coefficient. If the final coefficient for a metabolite is 0 then the metabolite is removed from the reaction. |
| build_reaction_string | use_metabolite_names=False (bool) | str | Generate a human readable reaction string |
| build_reaction_from_string | reaction_str (string) verbose (bool) fwd_arrow (re.compile) rev_arrow (re.compile) reversible_arrow (re.compile) term_split (string) |
None | Builds reaction from reaction equation reaction_str using parser Takes a string and using the specifications supplied in the optional arguments infers a set of metabolites, metabolite compartments and stoichiometries for the reaction. It also infers the reversibility of the reaction from the reaction arrow. |
| knock_out | None | None | Knockout reaction by setting its bounds to zero. |
# create new reactions
# cobra.Reaction(id=...)
new_rxns = [cobra.Reaction(id=f"R{i}", name=f"reaction No.{i}", lower_bound=0, upper_bound=1000) for i in range(1, 10)]
# add reactions to a model
model_copy.add_reactions(new_rxns)
print(model_copy.reactions[-10:])
print(f"Now the model has {len(model_copy.reactions)} reactions")
# remove reactions from a model
model_copy.remove_reactions(new_rxns)
print(f"Now the model has {len(model_copy.reactions)} reactions")
[<Reaction new_rxn_1 at 0x7fcd71787210>, <Reaction R1 at 0x7fcd71405890>, <Reaction R2 at 0x7fcd71406050>, <Reaction R3 at 0x7fcd71406010>, <Reaction R4 at 0x7fcd71405f50>, <Reaction R5 at 0x7fcd71405dd0>, <Reaction R6 at 0x7fcd71406850>, <Reaction R7 at 0x7fcd71406a10>, <Reaction R8 at 0x7fcd71406bd0>, <Reaction R9 at 0x7fcd71406d90>]
Now the model has 105 reactions
Now the model has 96 reactions
# get a reaction from a model
print(model.reactions[0])
print(model.reactions.get_by_id("ACALDt"),"\n")
# reactions attributes
rxn = model.reactions.get_by_id("ACALD")
print(f"ID: {rxn.id}\n\
Name: {rxn.name}\n\
Lower bound: {rxn.lower_bound}\n\
Upper bound: {rxn.upper_bound}\n\
Subsystem: {rxn.subsystem}\n\
Metabolites: {rxn.metabolites}\n\
Genes: {rxn.genes}\n\
Gene-reaction rule: {rxn.gene_reaction_rule}")
ACALD: acald_c + coa_c + nad_c <=> accoa_c + h_c + nadh_c
ACALDt: acald_e <=> acald_c
ID: ACALD
Name: acetaldehyde dehydrogenase (acetylating)
Lower bound: -1000.0
Upper bound: 1000.0
Subsystem:
Metabolites: {<Metabolite acald_c at 0x7fcd716c9850>: -1.0, <Metabolite coa_c at 0x7fcd7170a690>: -1.0, <Metabolite nad_c at 0x7fcd7171f7d0>: -1.0, <Metabolite accoa_c at 0x7fcd716cb4d0>: 1.0, <Metabolite h_c at 0x7fcd7171bad0>: 1.0, <Metabolite nadh_c at 0x7fcd7171fb50>: 1.0}
Genes: frozenset({<Gene b0351 at 0x7fcd71d98cd0>, <Gene b1241 at 0x7fcd84449150>})
Gene-reaction rule: b0351 or b1241
rxn.build_reaction_string()
'acald_c + coa_c + nad_c <=> accoa_c + h_c + nadh_c'
new_rxn = cobra.Reaction("dummy_rxn")
model_copy.add_reactions([new_rxn])
new_rxn
| Reaction identifier | dummy_rxn |
| Name | |
| Memory address | 0x7fcd7153cd50 |
| Stoichiometry |
--> --> |
| GPR | |
| Lower bound | 0.0 |
| Upper bound | 1000.0 |
# build a reaction by a human readable reaction string
new_rxn.build_reaction_from_string("acald_c + coa_c + nad_c --> accoa_c + h_c + nadh_c")
new_rxn
| Reaction identifier | dummy_rxn |
| Name | |
| Memory address | 0x7fcd7153cd50 |
| Stoichiometry |
acald_c + coa_c + nad_c --> accoa_c + h_c + nadh_c Acetaldehyde + Coenzyme A + Nicotinamide adenine dinucleotide --> Acetyl-CoA + H+ + Nicotinamide adenine dinucleotide - reduced |
| GPR | |
| Lower bound | 0 |
| Upper bound | 1000.0 |
# assign new values to the reaction attributes
new_rxn.name = "R123"
new_rxn.lower_bound, new_rxn.upper_bound = -1, 30
new_rxn.subsystem = "new subsystem"
new_rxn
| Reaction identifier | dummy_rxn |
| Name | R123 |
| Memory address | 0x7fcd7153cd50 |
| Stoichiometry |
acald_c + coa_c + nad_c <=> accoa_c + h_c + nadh_c Acetaldehyde + Coenzyme A + Nicotinamide adenine dinucleotide <=> Acetyl-CoA + H+ + Nicotinamide adenine dinucleotide - reduced |
| GPR | |
| Lower bound | -1 |
| Upper bound | 30 |
# knock out a reaction
new_rxn.knock_out()
new_rxn.bounds
(0, 0)
Metabolites (cobra.core.metabolite.Metabolite)#
attribute:#
| Attribute | Type | Description |
|---|---|---|
| id | str | the identifier to associate with the metabolite |
| formula | str | Chemical formula (e.g. H2O) |
| name | str | A human readable name. |
| charge | float | The charge number of the metabolite |
| compartment | str or None | Compartment of the metabolite. |
| elements | dict | Dictionary of elements as keys and their count in the metabolite as integer. |
| shadow_price | float | The shadow price in the most recent solution. |
method:#
| Method | Parameters | Return type | Description |
|---|---|---|---|
| remove_from_model | destructive (bool) | None | Removes the association from self.model |
| summary | solution=None (cobra.Solution) threshold=0.01 (float) fva=None (pandas.DataFrame or float) names=False (bool) float_format='{:.3g}'.format (callable) |
cobra.MetaboliteSummary | Create a summary of the producing and consuming fluxes. This method requires the model for which this metabolite is a part to be solved. |
# create a new metabolite
new_met = cobra.Metabolite(id="met_c", name="new metabolite", formula="C100H200", compartment="c")
print(new_met.elements)
new_met
{'C': 100, 'H': 200}
| Metabolite identifier | met_c |
| Name | new metabolite |
| Memory address | 0x7fcd71419990 |
| Formula | C100H200 |
| Compartment | c |
| In 0 reaction(s) |
# add a metabolite to a reaction
new_rxn.add_metabolites({
new_met: 1
})
new_rxn
| Reaction identifier | dummy_rxn |
| Name | R123 |
| Memory address | 0x7fcd7153cd50 |
| Stoichiometry |
acald_c + coa_c + nad_c --> accoa_c + h_c + met_c + nadh_c Acetaldehyde + Coenzyme A + Nicotinamide adenine dinucleotide --> Acetyl-CoA + H+ + new metabolite + Nicotinamide adenine dinucleotide - reduced |
| GPR | |
| Lower bound | 0 |
| Upper bound | 0 |
# remove a metabolite from a reaction
new_rxn.subtract_metabolites({
new_met: 1
})
new_rxn
| Reaction identifier | dummy_rxn |
| Name | R123 |
| Memory address | 0x7fcd7153cd50 |
| Stoichiometry |
acald_c + coa_c + nad_c --> accoa_c + h_c + nadh_c Acetaldehyde + Coenzyme A + Nicotinamide adenine dinucleotide --> Acetyl-CoA + H+ + Nicotinamide adenine dinucleotide - reduced |
| GPR | |
| Lower bound | 0 |
| Upper bound | 0 |
# remove metabolite from a model
model_copy.remove_metabolites([model_copy.metabolites.get_by_id("met_c")])
# get a metabolite from a model
print(model.metabolites[0])
print(model.metabolites.get_by_id("mal__L_c"),"\n")
# metabolites property
met = model.metabolites.get_by_id("mal__L_c")
print(f"ID: {met.id}\n\
Name: {met.name}\n\
Formula: {met.formula}\n\
Charge: {met.charge}\n\
Compartment: {met.compartment}\n\
Reactions: {met.reactions}\n")
13dpg_c
mal__L_c
ID: mal__L_c
Name: L-Malate
Formula: C4H4O5
Charge: -2
Compartment: c
Reactions: frozenset({<Reaction MDH at 0x7fcd715d6a10>, <Reaction ME1 at 0x7fcd715d7750>, <Reaction MALS at 0x7fcd715d4f50>, <Reaction ME2 at 0x7fcd715e4e90>, <Reaction FUM at 0x7fcd717a5990>, <Reaction MALt2_2 at 0x7fcd715d77d0>})
met.summary()
mal__L_c
C4H4O5
Producing Reactions
| Percent | Flux | Reaction | Definition |
|---|---|---|---|
| 100.00% | 5.064 | FUM | fum_c + h2o_c <=> mal__L_c |
Consuming Reactions
| Percent | Flux | Reaction | Definition |
|---|---|---|---|
| 100.00% | -5.064 | MDH | mal__L_c + nad_c <=> h_c + nadh_c + oaa_c |
Genes (cobra.core.gene.Gene)#
attribute:#
| Attribute | Type | Description |
|---|---|---|
| id | str | The identifier to associate the gene with |
| name | str | A longer human readable name for the gene |
| functional | bool | Indicates whether the gene is functional. If it is not functional then it cannot be used in an enzyme complex nor can its products be used. |
method:#
| Method | Parameters | Return type | Description |
|---|---|---|---|
| knock_out | None | None | Knockout gene by marking it as non-functional and setting all associated reactions bounds to zero. |
# create a new gene
new_gen = cobra.Gene(id="404", name="new gene")
print(new_gen.id, new_gen.name)
404 new gene
# get a gene
gene = model_copy.genes.get_by_id("b1241")
print(gene.id)
gene = model_copy.genes[10]
gene
b1241
| Gene identifier | b0726 |
| Name | sucA |
| Memory address | 0x7fcd7155fbd0 |
| Functional | True |
| In 1 reaction(s) | AKGDH |
# related rxn
gene.reactions
frozenset({<Reaction AKGDH at 0x7fcd7157ab50>})
# check boolean relationship
list(gene.reactions)[0].gene_reaction_rule
'b0726 and b0116 and b0727'
# add a gene to a model
print(f"Number of genes : {len(model_copy.genes)}")
list(gene.reactions)[0].gene_reaction_rule = f"{list(gene.reactions)[0].gene_reaction_rule} and 404"
print(f"Number of genes : {len(model_copy.genes)}")
Number of genes : 137
Number of genes : 138
# They are not the same object!!
model_copy.genes.get_by_id("404") == new_gen
print(id(model_copy.genes.get_by_id("404")), id(new_gen))
140520346671312 140521287593104
model_copy.reactions[0].gene_reaction_rule = f"{model_copy.reactions[0].gene_reaction_rule} or 404"
print(model_copy.genes.get_by_id("404").reactions)
frozenset({<Reaction ACALD at 0x7fcd71579650>, <Reaction AKGDH at 0x7fcd7157ab50>})
# knocking out a gene
model_copy.genes.get_by_id("404").knock_out()
# The related reactions might be influenced after you knock out a gene
for i in model_copy.genes.get_by_id("404").reactions:
print(i)
print(i.gene_reaction_rule)
print(f"{i.id} is functional? {i.functional}")
print()
ACALD: acald_c + coa_c + nad_c <=> accoa_c + h_c + nadh_c
b0351 or b1241 or 404
ACALD is functional? True
AKGDH: akg_c + coa_c + nad_c --> co2_c + nadh_c + succoa_c
b0726 and b0116 and b0727 and 404
AKGDH is functional? False
Configuration#
reference: https://cobrapy.readthedocs.io/en/stable/configuration.html
# The object has the following attributes which you can inspect but also change as desired.
cobra_config = cobra.Configuration()
cobra_config.bounds
(-1000.0, 1000.0)
# change default bounds
cobra_config.bounds = -10, 30
cobra.Reaction("R1", lower_bound=None)
| Reaction identifier | R1 |
| Name | |
| Memory address | 0x7fcd714182d0 |
| Stoichiometry |
<=> <=> |
| GPR | |
| Lower bound | -10 |
| Upper bound | 30 |
# change default solver
cobra_config.solver = "glpk_exact"
new_model = load_model("textbook")
Using context manager#
with model:
model.reactions[10].bounds = (-2000, 2000)
model.reactions[9].knock_out()
print(model.reactions[10].bounds)
print(model.reactions[9].bounds)
new_rxn2 = cobra.Reaction("r2")
new_met2 = cobra.Metabolite("m2")
model.add_metabolites([new_met2])
model.add_reactions([new_rxn2])
new_rxn2.build_reaction_from_string("m2 -->")
print(model.reactions.r2)
print(f"The model has {len(model.reactions)} reactions")
print(f"The model has {len(model.metabolites)} metabolites")
(-2000, 2000)
(0, 0)
r2: m2 -->
The model has 96 reactions
The model has 73 metabolites
# all the changes are reverted
print(model.reactions[10].bounds)
print(model.reactions[9].bounds)
try:
print(model.reactions.new_rxn2)
except:
print("Cannot find new_rxn2 in the model")
print(f"The model has {len(model.reactions)} reactions")
print(f"The model has {len(model.metabolites)} metabolites")
(8.39, 1000.0)
(-1000.0, 1000.0)
Cannot find new_rxn2 in the model
The model has 95 reactions
The model has 72 metabolites
Flux balance analysis(FBA)#
# The LP problem of the model
print(model.objective)
Maximize
1.0*Biomass_Ecoli_core - 1.0*Biomass_Ecoli_core_reverse_2cdba
# get a dict of {Reaction: objective_coefficient}
from cobra.util.solver import linear_reaction_coefficients
linear_reaction_coefficients(model)
{<Reaction Biomass_Ecoli_core at 0x7fcd71766850>: 1.0}
# modify the model objective
with model:
model.objective = model.reactions.TPI
print(f"obj value: {model.slim_optimize()}\n")
model.objective = {model.reactions.TPI: 2,
model.reactions.ACALD: 1}
# after running FBA, the flux of reaction can be obtained by:
model.optimize()
print(f"TPI Flux: {model.reactions.get_by_id('TPI').flux}\nACALD Flux: {model.reactions.get_by_id('ACALD').flux}\nobj value: {model.slim_optimize()}\n")
model.objective = "ACONTa"
print(f"obj value: {model.slim_optimize()}\n")
obj value: 10.000000000000034
TPI Flux: 10.000000000000002
ACALD Flux: 0.0
obj value: 20.000000000000004
obj value: 19.999999999999982
# Running FBA
sol = model.optimize()
sol
| fluxes | reduced_costs | |
|---|---|---|
| ACALD | 0.000000 | 3.864368e-18 |
| ACALDt | 0.000000 | -0.000000e+00 |
| ACKr | 0.000000 | -0.000000e+00 |
| ACONTa | 6.007250 | 0.000000e+00 |
| ACONTb | 6.007250 | 0.000000e+00 |
| ... | ... | ... |
| TALA | 1.496984 | -1.586585e-17 |
| THD2 | 0.000000 | -2.546243e-03 |
| TKT1 | 1.496984 | -1.914278e-17 |
| TKT2 | 1.181498 | 3.173170e-17 |
| TPI | 7.477382 | 0.000000e+00 |
95 rows × 2 columns
# attributes (analysis results) of a cobra.Solution object
sol.objective_value, sol.status
(0.8739215069684306, 'optimal')
# attributes (analysis results) of a cobra.Solution object
sol.to_frame().fluxes
ACALD 0.000000
ACALDt 0.000000
ACKr 0.000000
ACONTa 6.007250
ACONTb 6.007250
...
TALA 1.496984
THD2 0.000000
TKT1 1.496984
TKT2 1.181498
TPI 7.477382
Name: fluxes, Length: 95, dtype: float64
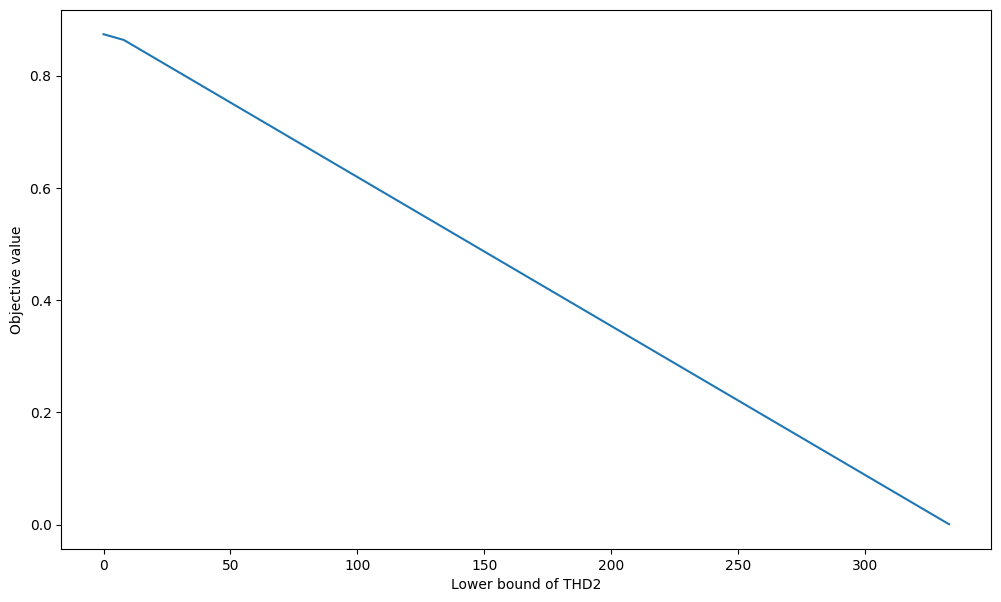
import numpy as np
import matplotlib.pyplot as plt
plt.figure(figsize=(12, 7))
with model:
X = np.arange(0, 1000)
Y = np.zeros(X.shape)
for i, x in enumerate(X):
THD2 = model.reactions.get_by_id("THD2")
THD2.lower_bound = x
Y[i] = model.slim_optimize()
plt.xlabel("Lower bound of THD2")
plt.ylabel("Objective value")
plt.plot(X, Y)
%%timeit
model.slim_optimize()
192 µs ± 6.73 µs per loop (mean ± std. dev. of 7 runs, 10,000 loops each)
%%timeit
model.optimize()
1.43 ms ± 17.4 µs per loop (mean ± std. dev. of 7 runs, 1,000 loops each)
model.summary()
Objective
1.0 Biomass_Ecoli_core = 0.8739215069684297
Uptake
| Metabolite | Reaction | Flux | C-Number | C-Flux |
|---|---|---|---|---|
| glc__D_e | EX_glc__D_e | 10 | 6 | 100.00% |
| nh4_e | EX_nh4_e | 4.765 | 0 | 0.00% |
| o2_e | EX_o2_e | 21.8 | 0 | 0.00% |
| pi_e | EX_pi_e | 3.215 | 0 | 0.00% |
Secretion
| Metabolite | Reaction | Flux | C-Number | C-Flux |
|---|---|---|---|---|
| co2_e | EX_co2_e | -22.81 | 1 | 100.00% |
| h2o_e | EX_h2o_e | -29.18 | 0 | 0.00% |
| h_e | EX_h_e | -17.53 | 0 | 0.00% |
model.metabolites.get_by_id("atp_c").summary()
atp_c
C10H12N5O13P3
Producing Reactions
| Percent | Flux | Reaction | Definition |
|---|---|---|---|
| 66.58% | 45.51 | ATPS4r | adp_c + 4.0 h_e + pi_c <=> atp_c + h2o_c + 3.0 h_c |
| 23.44% | 16.02 | PGK | 3pg_c + atp_c <=> 13dpg_c + adp_c |
| 2.57% | 1.758 | PYK | adp_c + h_c + pep_c --> atp_c + pyr_c |
| 7.41% | 5.064 | SUCOAS | atp_c + coa_c + succ_c <=> adp_c + pi_c + succoa_c |
Consuming Reactions
| Percent | Flux | Reaction | Definition |
|---|---|---|---|
| 12.27% | -8.39 | ATPM | atp_c + h2o_c --> adp_c + h_c + pi_c |
| 76.46% | -52.27 | Biomass_Ecoli_core | 1.496 3pg_c + 3.7478 accoa_c + 59.81 atp_c + 0.361 e4p_c + 0.0709 f6p_c + 0.129 g3p_c + 0.205 g6p_c + 0.2557 gln__L_c + 4.9414 glu__L_c + 59.81 h2o_c + 3.547 nad_c + 13.0279 nadph_c + 1.7867 oaa_c + 0.5191 pep_c + 2.8328 pyr_c + 0.8977 r5p_c --> 59.81 adp_c + 4.1182 akg_c + 3.7478 coa_c + 59.81 h_c + 3.547 nadh_c + 13.0279 nadp_c + 59.81 pi_c |
| 0.33% | -0.2235 | GLNS | atp_c + glu__L_c + nh4_c --> adp_c + gln__L_c + h_c + pi_c |
| 10.94% | -7.477 | PFK | atp_c + f6p_c --> adp_c + fdp_c + h_c |
Flux variability analysis(FVA)#
from cobra.flux_analysis import flux_variability_analysis as fva
sol = fva(model, fraction_of_optimum=0.8)
sol
| minimum | maximum | |
|---|---|---|
| ACALD | -5.084741 | 0.000000 |
| ACALDt | -5.084741 | 0.000000 |
| ACKr | -7.564396 | 0.000000 |
| ACONTa | 0.754299 | 10.128462 |
| ACONTb | 0.754299 | 10.128462 |
| ... | ... | ... |
| TALA | -0.154536 | 9.249088 |
| THD2 | 0.000000 | 69.950657 |
| TKT1 | -0.154536 | 9.249088 |
| TKT2 | -0.466373 | 8.996699 |
| TPI | -0.069595 | 9.304568 |
95 rows × 2 columns
model.summary(fva=0.8)
Objective
1.0 Biomass_Ecoli_core = 0.873921506968433
Uptake
| Metabolite | Reaction | Flux | Range | C-Number | C-Flux |
|---|---|---|---|---|---|
| glc__D_e | EX_glc__D_e | 10 | [8.093; 10] | 6 | 100.00% |
| nh4_e | EX_nh4_e | 4.765 | [3.812; 6.355] | 0 | 0.00% |
| o2_e | EX_o2_e | 21.8 | [14.31; 29.44] | 0 | 0.00% |
| pi_e | EX_pi_e | 3.215 | [2.572; 3.215] | 0 | 0.00% |
Secretion
| Metabolite | Reaction | Flux | Range | C-Number | C-Flux |
|---|---|---|---|---|---|
| ac_e | EX_ac_e | 0 | [-7.564; 0] | 2 | 0.00% |
| acald_e | EX_acald_e | 0 | [-5.085; 0] | 2 | 0.00% |
| akg_e | EX_akg_e | 0 | [-2.86; 0] | 5 | 0.00% |
| co2_e | EX_co2_e | -22.81 | [-30.25; -10.81] | 1 | 100.00% |
| etoh_e | EX_etoh_e | 0 | [-4.429; 0] | 2 | 0.00% |
| for_e | EX_for_e | 0 | [-19.06; 0] | 1 | 0.00% |
| glu__L_e | EX_glu__L_e | 0 | [-2.542; 0] | 5 | 0.00% |
| h2o_e | EX_h2o_e | -29.18 | [-35.34; -15.91] | 0 | 0.00% |
| h_e | EX_h_e | -17.53 | [-33.08; -14.02] | 0 | 0.00% |
| lac__D_e | EX_lac__D_e | 0 | [-4.29; 0] | 3 | 0.00% |
| pyr_e | EX_pyr_e | 0 | [-5.085; 0] | 3 | 0.00% |
| succ_e | EX_succ_e | 0 | [-3.348; 0] | 4 | 0.00% |
model.metabolites.get_by_id("atp_c").summary(fva=0.8)
atp_c
C10H12N5O13P3
Producing Reactions
| Percent | Flux | Range | Reaction | Definition |
|---|---|---|---|---|
| 66.58% | 45.51 | [27.44; 69.45] | ATPS4r | adp_c + 4.0 h_e + pi_c <=> atp_c + h2o_c + 3.0 h_c |
| 23.44% | 16.02 | [8.767; 18.14] | PGK | 3pg_c + atp_c <=> 13dpg_c + adp_c |
| 2.57% | 1.758 | [0; 39.63] | PYK | adp_c + h_c + pep_c --> atp_c + pyr_c |
| 7.41% | 5.064 | [0; 9.374] | SUCOAS | atp_c + coa_c + succ_c <=> adp_c + pi_c + succoa_c |
Consuming Reactions
| Percent | Flux | Range | Reaction | Definition |
|---|---|---|---|---|
| 0.00% | 0 | [0; 7.564] | ACKr | ac_c + atp_c <=> actp_c + adp_c |
| 0.00% | 0 | [-34.32; 0] | ADK1 | amp_c + atp_c <=> 2.0 adp_c |
| 12.27% | -8.39 | [-42.71; -8.39] | ATPM | atp_c + h2o_c --> adp_c + h_c + pi_c |
| 76.46% | -52.27 | [-52.27; -41.82] | Biomass_Ecoli_core | 1.496 3pg_c + 3.7478 accoa_c + 59.81 atp_c + 0.361 e4p_c + 0.0709 f6p_c + 0.129 g3p_c + 0.205 g6p_c + 0.2557 gln__L_c + 4.9414 glu__L_c + 59.81 h2o_c + 3.547 nad_c + 13.0279 nadph_c + 1.7867 oaa_c + 0.5191 pep_c + 2.8328 pyr_c + 0.8977 r5p_c --> 59.81 adp_c + 4.1182 akg_c + 3.7478 coa_c + 59.81 h_c + 3.547 nadh_c + 13.0279 nadp_c + 59.81 pi_c |
| 0.33% | -0.2235 | [-34.5; -0.1788] | GLNS | atp_c + glu__L_c + nh4_c --> adp_c + gln__L_c + h_c + pi_c |
| 10.94% | -7.477 | [-43.1; 0] | PFK | atp_c + f6p_c --> adp_c + fdp_c + h_c |
| 0.00% | 0 | [-34.32; 0] | PPCK | atp_c + oaa_c --> adp_c + co2_c + pep_c |
| 0.00% | 0 | [-34.32; 0] | PPS | atp_c + h2o_c + pyr_c --> amp_c + 2.0 h_c + pep_c + pi_c |
### pFBA
from cobra.flux_analysis import pfba
sol = pfba(model)
sol
| fluxes | reduced_costs | |
|---|---|---|
| ACALD | 0.000000e+00 | -2.000000 |
| ACALDt | 0.000000e+00 | 2.000000 |
| ACKr | -2.484585e-14 | 2.000000 |
| ACONTa | 6.007250e+00 | -2.000000 |
| ACONTb | 6.007250e+00 | -2.000000 |
| ... | ... | ... |
| TALA | 1.496984e+00 | -2.000000 |
| THD2 | 0.000000e+00 | 3.822222 |
| TKT1 | 1.496984e+00 | -2.000000 |
| TKT2 | 1.181498e+00 | -2.000000 |
| TPI | 7.477382e+00 | -2.000000 |
95 rows × 2 columns
sol.objective_value
518.4220855176067
# the objective value is the sum of fluxes
sol.fluxes.abs().sum()
518.4220855176065